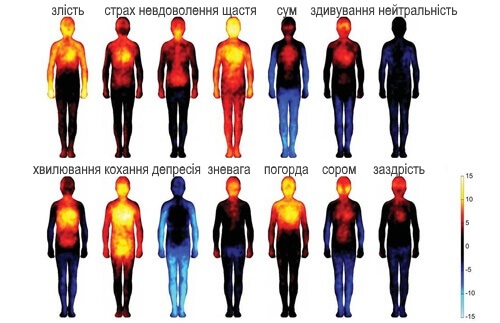
Як працюють «гормони щастя»
Всі ми чули
вислів «гормони щастя». Невже омріяне щастя — результат хімічних процесів? Ми
вирішили з’ясувати, що це за гормони, і як вони працюють.
Гормони щастя — що це?
«Гормонами
щастя» прийнято називати серотонін, ендорфін і дофамін. Серотонін виробляється
в шишкоподібної залозі головного мозку. Як хімічна сполука, серотонін
відноситься до того ж ряду алкалоїдів індолу, що і психоделічні наркотики
начебто ЛСД-25, псилоцибіна, ДМТ і буфотенина.
Він був
відкритий в 1935 році італійським фармакологом Вітторіо Ерспамером, який назвав
це речовиною, що скорочує гладку мускулатуру, ентерамином.
Через 13
років у клініці Влівленда Моріс Раппорт з колегами виявив судинозвужувальну
речовину в сироватці крові, яка було названа серотоніном. В 1952 році було
доведено, що серотонін і ентерамин — це одна і та ж речовина, а ще через рік
серотонін був виявлений в головному мозку.
Ендорфіни
представляють із себе групу хімічних сполук, за способом дії схожих з опіатами.
Вони утворюються виробляється гіпофізом речовини беталіпотрофіну. Серотонін
утворюється з амінокислоти триптофану.
Нейромедіатр
дофамін виробляється в головному мозку і наднирниках. Він відіграє велику роль
в «системі винагороди» мозку і виробляється під час позитивного для людини
досвіду.

Чи дійсно серотонін робить людину щасливою?
З
серотоніном все не так просто. Уявлення про матерію як про панацею від смутку і
депресії невірно. В даний час вплив серотоніну на поведінку людини ще
недостатньо вивчено. Він впливає по-різному на різні ділянки мозку, і цей вплив
нерівномірно і залежить від взаємодій з іншими гормонами.
Найменша
концентрація серотоніну в корі та мозку, найбільша – активує ретикулярну
систему, де він грає важливу роль в циклі сну-неспання. Коли рівень серотоніну
тут підвищується, мозок занурюється в глибокий сон.
Дослідження,
що проводилися в психіатричних клініках, показали, що надмірний вміст
серотоніну виявляється в мозку душевнохворих.
Сьогодні вже
доведено зв’язок між дефіцитом серотоніну і агресією. Це пов’язаність язано з
тим, що серотонін контролює інші трансмітери і «вирішує», пропускати той чи
інший сигнал у мозок. Коли серотоніну недостатньо, цей контроль слабшає і
адреналінові реакції включають в мозку механізми паніки, агресивності навіть
тоді, коли видимих причин для цього немає.
Профіцит
серотоніну також може негативно позначитися на здоров ї та поведінці людини.
Так, підвищений вміст серотоніну спостерігається при аутичних станах. Нездатність
рецепторів зв’язку зв’язати велику кількість серотоніну також зазначалося у
людей, страждаючих хворобою Альцгеймера.
Щасливі гени
Останні
дослідження довели, що для належного рівня щастя людині потрібні відповідні
гени, а точніше, так званий ген 5-HTTLPR, який відповідає за транспортування
вже згадуваного серотоніну. Для того, щоб це довести, вчені Лондонської
медичної школи та школи економіки опитали кілька тисяч чоловік. Результати
підтвердили припущення: волонтери, у яких було дві копії гена щастя від обох
батьків, виявилися оптимістами, не схильними до депресії. Результати
дослідження були опубліковані Ян-Еммануелем де Неві в журналі Journal of Human
Genetics.
Вчений також
підкреслив, що незабаром можуть бути знайдені й інші «щасливі гени». Однак
Ян-Еммануель не стверджує, що при несприятливих умовах люди з «щасливими
генами» не зазнають депресії. Гени генами, але реакції організму на негативні
фактори ніхто не відміняв.
Щаслива страва
Як ми вже
знаємо, серотонін утворюється з амінокислоти триптофану. Потреба в ній може
задовольнятися лише вступом її з-зовні, тому життєво важливо споживати
«правильні» продукти, що сприяють виробленню серотоніну і ендорфінів. Триптофан
— компонент харчових білків, що містяться в соєвих бобах, банани, фініки, горіхи,
гриби, вівсі.
Благотворно
на вироблення серотоніну також позначається споживання шоколаду, картоплі,
авокадо, кінзи і іншої зелені, молока, цитрусових, цільнозернового хліба і
сиру. Рівень ендорфінів також підвищується при прослуховуванні музики та від занять
сексом.
Важливо пам
” ятати про те, що триптофан перетворюється в серотонін в шишкоподібної залозі
тільки при сонячному світлі, тому не потрібно нехтувати прогулянками та
відпочинком на свіжому повітрі. Зимова сезонна депресія — із-за нестачі
природного світла. Також виробленню ендорфінів сприяють заняття спортом. Людина
може в прямому сенсі, «добігти до щастя».
Що шкодить гормону щастя?
Негативно
позначається на вироблення «гормонів щастя» прийом будь-яких наркотичних
речовин. Із-за їх вживання порушуються медіаторні мозкові зв’язки. По суті,
синтетичні психоактивні речовини обманюють мозок, видаючи себе за серотонін.
Швидкий викид ендорфінів у плазму крові і мозок після вживання стимуляторів
провокує звикання до цього рівня. Згодом організм перестає виробляти необхідні
гормони, що вкрай негативно позначається на хімічному балансі організму,
призводить до депресії та інших негативних наслідків